编者按:量子化学中的哈密顿量计算一直是制约药物筛选与新材料设计效率的瓶颈。近期,微软研究院科学智能中心、耶鲁大学及加州大学戴维斯分校联合研究提出的全新损失函数 WALoss 和深度学习框架 WANet,为这一难题提供了创新的解决方案。通过优化计算过程,WANet 显著提高了哈密顿量预测的精度与效率,尤其在大分子系统中展示了强大的扩展能力。搭配 WALoss,WANet 则能够准确预测分子能量,并有效捕捉分子的电子分布,成为化学、材料科学等领域多维分析的可靠工具。WANet 与 WALoss 的提出有望推动量子化学的广泛应用,助力高通量药物筛选、新材料设计和基础科学研究的创新进展。
近年来,密度泛函理论(Density Functional Theory, DFT)凭借其高效性和准确性,已成为量子化学与材料科学研究中的核心工具,广泛应用于预测分子电子结构、光谱特性及分子动力学模拟。DFT 的核心在于求解 Kohn-Sham 方程,通过构建 Kohn-Sham 哈密顿量获取分子系统的关键量子态信息,如 HOMO(最高占据分子轨道)和 LUMO(最低未占据分子轨道)能量、HOMO-LUMO 能隙以及总能量和光谱特性,这些信息为研究分子振动、反应路径和结构优化提供了重要基础。
然而,传统自洽场方法(Self-consistent field, SCF)在构建 Kohn-Sham 哈密顿量时具有 O(N³) 到 O(N⁴) 的计算复杂度(N 为电子数),在原子数量较多的大分子系统中,计算资源需求呈指数增长,导致应用受限。现有深度学习方法虽在小分子系统上有所突破,但在大分子系统上往往因数据稀缺和误差积累导致非物理性预测结果,从而显著降低了模型的可靠性与适用性。
对此,微软研究院科学智能中心、耶鲁大学及加州大学戴维斯分校合作提出了全新的损失函数波函数对齐损失 WALoss(Wavefunction Alignement Loss) 和现代化深度学习架构 WANet [1], 为提升 Kohn-Sham 哈密顿量的可扩展性与预测精度提供了全新的解决方案。这项研究在分子系统建模方面取得了显著突破,助力破解大规模分子系统的计算难题。相关研究成果 Enhancing the Scalability and Applicability of Kohn-Sham Hamiltonians for Molecular Systems (opens in new tab) 已被全球机器学习领域顶会 ICLR 2025 评为 Spotlight 研究。
传统方法在大分子哈密顿量预测中存在严重偏差?
为了初步验证现有网络在哈密顿量可扩展性上的不足,研究员们开发了一个全新的数据集 PubChemQH。该数据集基于 GPU 加速的密度泛函理论软件 MADFT [2],使用了 def2-tzvp 基组和 B3LYP 泛函,在128张 NVIDIA V100 显卡上进行了一个月的不间断计算生成。PubChemQH 数据集包含50,000个分子样本,样本分子结构中的原子数量范围从40到100,超过此前金标准数据集 QH9(仅包含不超过31个原子的分子)。这一大规模数据集的引入,为后续探索大分子系统中的可扩展性提供了基础。
在 PubChemQH 数据集上,研究员们发现,尽管传统深度学习方法在哈密顿量预测中表现出较低的平均误差,但其预测的物理性质(如总能量、波函数、HOMO、LUMO 等)偏差极大。其中,总能量的预测误差高达约10,000 kcal/mol,远远无法达到化学精度(通常要求误差在 1 kcal/mol 以内),严重影响了模型的实际可用性。
突破哈密顿量计算瓶颈
针对预测偏差大的问题,研究员们提出了全新的损失函数波函数对齐损失 WALoss 和现代化深度学习架构 WANet。WALoss 旨在通过对齐预测波函数与真实波函数的基,来提升模型预测的物理准确性。由于传统方法大多采用均方误差(MSE)或平均绝对误差(MAE)作为损失函数,这些损失函数仅对哈密顿矩阵上的每一项进行单独比较,所以无法有效捕捉哈密顿量在整体结构上的偏差,尤其在处理大分子时往往会失效。WALoss 可通过基变换对波函数进行对齐,使得预测的哈密顿量更加符合物理真实值,从而减少预测误差并显著提升 SCF 计算的收敛速度。
专门用于 Kohn-Sham 哈密顿量预测的现代化网络架构 WANet 则进一步提升了网络的计算效率。WANet 的构建灵感来源于等变图神经网络 SE(3)-Equivariant Networks,该网络能够自动捕捉分子的旋转、平移及对称性等物理特征,有效提高了模型对分子结构的理解能力。
在 WANet 的网络架构中,节点卷积层(Node Convolution Layer)承担着捕捉局部原子间复杂相互作用的任务。通过对局部原子邻域信息的图卷积操作,该层能够生成节点的高阶不可约表示,为下游的哈密顿量预测构建模块提供支持。具体地,WANet 将之前网络中常使用的 SO(3) 卷积改为了基于 SO(2) 的卷积神经网络,保证了网络等变的同时,大幅提升了计算效率。最后,哈密顿量预测模块则利用 Clebsch-Gordan 张量积技术,将高阶不可约表示进一步转化为完整的哈密顿量矩阵,并分别构建对角块和非对角块。

WANet 和 WALoss 实验验证
实验结果表明,WANet 和 WALoss 在 PubChemQH 数据集上取得了极大的性能提升。在 PubChemQH 数据集上(如表1),WANet 搭配 WALoss 后,系统能量预测误差从 QHNet 的65721.028 kcal/mol 降至47.193 kcal/mol,同时 SCF 迭代次数减少至82%,远低于传统方法的371%。
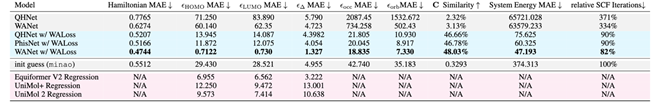
计算效率是深度学习在量子化学应用中的重要考量。WANet 在推理速度和 GPU 资源利用方面展现出显著优势。相比传统 DFT 方法,WANet 不仅减少了 SCF 迭代次数,还大大降低了总计算时间。GPU 内存占用也从 QHNet 的26.49 GB 降至15.86 GB,使得 WANet 更加适合高通量计算场景。

除了能量预测,WANet 在偶极矩(dipole moment)和电子空间范围(Electronic Spatial Extent, ESE)的预测中也表现优异。实验数据显示,WANet 搭配 WALoss 后在 Dipole Moment 预测中的误差仅为3.3928 D,显著优于其他基于深度学习的回归模型。同时,在 ESE 预测中,WANet 的误差为0.0076 a.u.,远低于传统方法。这一结果证明 WANet 不仅在能量预测方面具有优势,还能有效捕捉分子的空间电子分布,为分子性质的多维度分析提供了可靠工具。

WANet 还具有出色的扩展能力。在碳链延伸分子(CxH2x+2)的测试中,当碳原子数量超过 PubChemQH 数据集的范围(超过100个原子)时,WANet 依然保持了高精度预测,尤其在 LUMO 能量的预测中显著优于传统方法。这一结果表明,WANet 能够有效泛化到更大规模的分子系统。
未来应用潜力
WANet 和 WALoss 的提出为密度泛函理论的进一步发展注入了技术活力,也为高通量药物筛选、新材料设计及基础科学研究开辟了全新路径。随着更高质量数据集的引入和模型架构的不断优化,基于深度学习的哈密顿量预测方法有望在未来实现更广泛的应用,推动量子化学从理论走向实践,助力基础科学与工业技术的进步。
相关链接:
[1] https://openreview.net/forum?id=twEvvkQqPS
[2] Ju F, Wei X, Huang L, et al. Acceleration without disruption: DFT software as a service[J]. Journal of Chemical Theory and Computation, 2024, 20(24): 10838-10851.